Острый промиелоцитарный лейкоз


Содержание:
- Определение
- Причины
- Симптомы
- Диагностика
- Профилактика
Определение
Острый промиелоцитарный лейкоз (ОПЛ) является подтипом острого миелолейкоза (ОМЛ), рака крови и костного мозга. Составляет 5-10% случаев ОМЛ. Встречается во всех возрастных группах, но очень редко у детей в возрасте до 10 лет. Средний возраст больных – 38-40 лет, соотношение мужчин и женщин составляет 2:1.
Основными симптомами, связанными с геморрагическими проявлениями, наблюдаются у 90 % больных. Гепатомегалия и лимфаденопатия отмечается не более чем у 20% пациентов.
Причины
Истинная частота возникновения ОПЛ неизвестна, поскольку до недавнего времени в регистры заболеваемости острыми лейкозами ОПЛ вносили вместе с другими вариантами ОМЛ. Считается, что ОПЛ встречается в 5-15% всех случаев ОМЛ.
Существует несколько отличий в эпидемиологических характеристиках между острым миелоцитарным лейкозом (ОМЛ) и острым промиелоцитарным лейкозом, особенно это касается вероятности возникновения ОПЛ в зависимости от возраста.
Так, для ОМЛ вероятность возникновения постепенно увеличивается пропорционально возрасту до 55 лет, а затем отмечается резкий экспоненциальный рост заболеваемости. При ОПЛ эта закономерность не выявляется.
Хотя заболевание диагностируется во всех возрастных группах, но его частота крайне низка у больных в возрасте до 10 лет. В возрастной группе от 0 до 17 лет частота ОПЛ среди всех случаев ОМЛ составляет 3-4%.
В возрасте от 10 лет до 20 лет вероятность возникновения ОПЛ постепенно возрастает, затем наблюдается плато до возраста 60 лет, после чего вероятность возникновения заболевания снижается. Большинство случаев ОПЛ диагностируют в возрасте от 20 до 60 лет. Медиана возраста при диагностике ОПЛ составляет 38 лет.
Другой существенной эпидемиологической характеристикой ОПЛ является высокая частота встречаемости у больных из Латинской Америки – 24,3%, что значительно выше, чем у представителей других этнических групп. Однако особенностей клинического течения заболевания или каких-либо принципиальных биологических отличий у них не обнаруживается.
Также следует отметить, что частота встречаемости различных вариантов точек разрыва в гене PML (bcrl, bcr2, ЬсгЗ) отличается в зависимости от этнического происхождения. Например, отмечают большую частоту выявления (63- 75%) у больных латиноамериканского происхождения bcrl транскрипта, чем у больных в Италии – 59,4%, Испании – 56%, Великобритании – 61%, США – 54%.
Столь же высокие цифры по частоте определения первого варианта транскрипта отмечены и у больных из Азии – в Китае, например, встречаемость составляет 69%.
Полагают, что у больных из Латинской Америки, большинство из которых являются метисами, то есть потомками кавказоидов и американских аборигенов, иммигрировавших в Америку из восточной Азии почти 12 тысяч лет назад, сохранилась генетическая предрасположенность, свойственная азиатам, к более высокой частоте bcrl.
В последние годы все больше описывается случаев возникновения ОПЛ как вторичного лейкоза, связанного с предшествующей химиотерапией и облучением.
Большие многоцентровые исследования свидетельствуют о том, что вторичный ОПЛ в большинстве случаев возникает не позднее трех лет после завершения химиотерапии по поводу первичного онкологического заболевания ингибиторами топоизомеразы II (антрациклины, или митоксантрон, реже – этопозид).
У 57% больных первичной опухолью был рак молочной железы, далее следуют лимфомы, значительно реже лимфогранулематоз. Среднее время от завершения терапии по поводу первичной опухоли до момента диагностики вторичного ОПЛ составляет 24 месяца (от 15 месяцев до 8 лет).
По мере увеличения агрессивности химиотерапевтического воздействия увеличивается вероятность развития вторичного ОПЛ, как, впрочем, может уменьшаться и временной интервал от момента завершения химиотерапии и возникновения острого лейкоза.
Все вторичные ОПЛ были отнесены к классическому гипергранулярному варианту, во всех этих случаях выявлялась транслокация t (15; 17), результаты терапии при них не отличались от таковых при первичных ОПЛ. Все исследователи отмечают, что прогноз при вторичном ОПЛ столь же оптимистичный, как и при de novo в случаях, если использовать адекватную химиотерапевтическую тактику.
Интересно отметить, что описаны промиелоцитарные властные кризы хронического миелолейкоза (ХМЛ). В описываемых случаях промиелоцитарный бластный криз был диагностирован у больных с ранее доказанным хроническим миелолейкозом.
В одном случае диагноз промиелоцитарного криза подтверждается морфологически, иммунофенотипически и цитогенетически (FISH-исследование на t). В другом случае при властном кризе выявляют типичный маркер ОПЛ – транскрипт PML-RARa.
Симптомы
ОПЛ представляет собой четко очерченную нозологическую форму с настолько характерными клинико-лабораторными признаками (типичная морфология опухолевых клеток, тяжелый геморрагический синдром, гематомный тип кровоточивости, избыточно активированный фибринолиз, ДВС-синдром, обычно лейкопения), что диагноз порой можно установить, основываясь лишь на клинических проявлениях. При этом в клинической практике можно столкнуться и со случаями ОПЛ, которые протекают не столь драматично – отсутствуют проявления геморрагического синдрома, больные в течение нескольких месяцев могут наблюдаться по поводу лейкопении, умеренной тромбоцитопении.
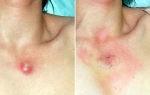
Проявления геморрагического синдрома (кровоточивость десен, повышенная травмируемость кожных покровов, синяки, петехии, нередко кровотечения из носа, желудочно-кишечного тракта) имеются у 90% пациентов на момент диагностики. Гепатоспленомегалия или лимфоаденопатия определяются менее чем у 20% пациентов.
У 80% больных манифестация заболевания характеризуется лейкопенией (медиана числа лейкоцитов составляет 1,8х109/л.), причем, чаще всего лейкопения обнаруживается у больных с гипергранулярным вариантом ОПЛ. Хотелось бы отметить, что если у больного в момент диагностики острого лейкоза определяется лейкопения менее 1х109/л.
, особенно в сочетании с гипофибриногенемией, то с большой долей вероятности можно предполагать промиелоцитарный вариант ОМЛ. У 15-20% пациентов, преимущественно с гипогранулярным вариантом заболевания, в дебюте болезни выявляется гиперлейкоцитоз (медиана 83х109/л.) с увеличением числа лейкоцитов до 150 и более тысяч.
У подавляющего числа больных (80-90%) определяется анемия, причем у половины из них концентрация гемоглобина составляет менее 100 г/л. У 75% больных содержание тромбоцитов снижается до 50х109/л. и ниже.
Лабораторные признаки диссеминированного внутрисосудистого свертывания крови и истощенного фибринолиза определяются у 80-90% пациентов.
Диагностика
Морфологические особенности опухолевых клеток при ОПЛ настолько характерны, что в большинстве случаев не требуется никаких дополнительных методов исследования для установления правильного диагноза.
С момента выделения ОПЛ в отдельную форму ОМЛ, все исследователи отмечали, что бластные клетки при ОПЛ прежде всего характеризуются значительным ядерным полиморфизмом и наличием крупной фиолетово-бурой зернистости, густо заполняющей цитоплазму.
Вне всякого сомнения, эти морфологические признаки свойственны классическому – гипергранулярному – варианту ОПЛ. Гипогранулярный вариант определяется в 15-20% всех случаев ОПЛ.
При обычной окраске в цитоплазме опухолевых клеток обнаруживается лишь несколько мелких гранул или они не выявляются вовсе, при этом все остальные признаки (клинические, цитогенетические) ОПЛ присутствуют.
Для ОПЛ исключительно важным является получение адекватного костномозгового пунктата, так как большинство форм характеризуется глубокой лейкопенией. Также высока вероятность того, что аспират костного мозга свернется, и диагностический материал не будет получен.
В большинстве случаев ОПЛ (90-95%) процент миелобластов составляет менее 30, в среднем 8-10. Основным опухолевым субстратом являются аномальные промиелоциты, в цитоплазме которых содержится множество грубых азурофильных гранул, плотность которых часто такова, что контуры ядра клетки не определяются.
Азурофильные гранулы обычно больше по размерам (иногда в несколько раз) и более яркие по окраске, чем в нормальных промиелоцитах. Часто при морфологическом анализе выявляется небольшая популяция промиелоцитов, которые содержат обильную, необычно нежную, ржавого цвета зернистость. Ядро клеток характеризуется исключительным разнообразием (и по размерам, и по форме, и по структуре).
Ядерный хроматин может быть нежным, «бластным» по структуре, или скомканным, грубым, неоднородным, как у незрелых моноцитов. Причем, клетки с моноцитоидным ядром содержат относительно редкие гранулы. Иногда может выявляться популяция клеток с очень необильной азурофильной зернистостью, но резко базофильной цитоплазмой, в которых ядро двухдольчатое или как-то необычно разделено на доли.
Размеры большинства таких клеток в два раза меньше, чем у нормального промиелоцита. Эти гипербазофильные промиелоциты чаще всего выявляются в незначительном количестве. Реже они могут составлять основную популяцию клеток периферической крови, при этом в костном мозге будут присутствовать классические гипергранулированные промиелоциты.
Гипербазофильные промиелоциты могут иногда напоминать микромегакариоциты, тем более что в этих клетках в ряде случаев описываются цитоплазматические выросты.
В 90-95% случаев гипергранулярного варианта ОПЛ в промиелоцитах обнаруживаются множественные тонкие палочки Ауэра, часто переплетенные между собой, напоминающие «вязанки хвороста». В некоторых случаях эти пучки палочек Ауэра легче выявляются на недокрашенных мазках, нежели на хорошо прокрашенных препаратах.
В редких случаях в промиелоцитах определяются крупные, круглые или овальные азурофильные включения, напоминающие таковые при синдроме Чегиака-Хигаси. В созревающих нейтрофилах при ОПЛ могут определяться диспластические признаки – малосегментированное ядро и уменьшение числа гранул в цитоплазме.
Мазок периферической крови в ряде случаев обнаруживает морфологические признаки разрушения эритроцитов вследствие диссеминированного внутрисосудистого свертывания (ДВС) – анизоцитоз, сфероцитоз. При микрогранулярном варианте ОПЛ зернистость в промиелоцитах очень нежная, менее плотная, иногда совсем отсутствует.
Ядро в клетках отличается значительной уродливостью – складчатое, дольчатое, напоминающее моноциты. Почти всегда можно найти клетки с характерными палочками Ауэра или подобными им включениями.
Интересен тот факт, что при тщательном микроскопическом исследовании опухолевых клеток ОПЛ могут быть выделены определенные морфологические признаки, позволяющие предположить наличие тех или иных цитогенетических аномалий, свойственных различным вариантам ОПЛ. Выделено 15 морфологических подтипов ОПЛ.
Оценивалась форма ядра (правильная – округлая или овальная; неправильная – бобовидная, двухлопастная, дольчатая), степень гранулированности цитоплазмы (гипергранулярная и гипогранулярная, азурофильные гранулы), наличие или отсутствие палочек Ауэра.
Также отдельно оценивалось присутствие базофильных гранул, гранул Чегиака-Хигаси, наличие пельгероподобных клеток. Анализ сделан на небольшом материале, (90 пациентов) и, конечно, не может служить однозначным руководством к определению цитогенетического варианта ОПЛ без проведения самого цитогенетического анализа.
Любая морфологическая оценка является в достаточной степени субъективной и, по-видимому, требует экспертного подхода.
Тем не менее, если при ОПЛ бластные клетки характеризуются правильной 4 формой ядра (не дольчатое, не двухлопастное) с четкими контурами, отсутствием пучков палочек Ауэра, мелкой зернистостью, если встречается большое количество пельгероподобных клеток, то можно предположить, что при данном ОПЛ будет выявлена транслокация t (15; 17) с химерным геном PLZF/RARoc.
Помимо столь подробного дифференцирования разных морфологических вариантов ОПЛ, существуют более простые способы выделения различных подтипов ОПЛ.
Итальянские исследователи предлагают обращать внимание на следующие формы: 1) классический гипергранулярный ОПЛ; 2) микрогранулярный ОПЛ; 3) ОПЛ с избытком базофилов; 4) эозинофильный лейкоз с маркером ОПЛ PML-RARa; 5) ОПЛ с базофильными гранулами; 6) морфологически подобный ОПЛ лейкоз без классических маркеров.
Обычная гистологическая характеристика ОПЛ (по данным трепанобиопсии костного мозга) включает в себя резко гиперклеточный костный мозг, представленный, в основном, атипичными промиелоцитами с единичными островками остаточного нормального гемопоэза. Редко встречаются скопления эритробластов, единичные мегакариоциты.
Аномальные промиелоциты характеризуются интенсивно окрашенной эозинофильной цитоплазмой при окраске гематоксилин-эозином. В цитоплазме четко видна зернистость, которая может напоминать таковую у тучных клеток или эозинофилов. Причем, даже в гистологическом препарате могут быть видны палочки Ауэра.
Границы цитоплазмы у опухолевых клеток чаще всего неправильные или плохо определяются. При малом увеличении аномальные промиелоциты могут напоминать также плазматические клетки.
При ОПЛ в костном мозге отмечается резкое увеличение васкуляризации и такой показатель, как «плотность микрососудов» (число сосудов на одно поле зрения при увеличении 600х), существенно превышает контрольные показатели: 7 в сравнении с 2,4 сосудов на одно поле зрения (р
Источник: http://med36.com/ill/2383
Острый промиелоцитарный лейкоз: продолжительность жизни, прогноз
Содержание
Острый промиелоцетарный лейкоз относится к опухолевым заболеваниям кровеносной системы и является разновидностью миелоидного лейкоза (ОМЛ). Был описан в 1957 г. и заслужил плохую репутацию.
Но современная медицина позволяет жить с таким заболеванием: 12 лет без рецидива.
При стандартном проявлении, успешность лечения составляет более 70%, что превышает средний процент благоприятного исхода среди патологий данной группы.
Причины и механизмы воздействия
Синтез белых клеток (гранулоцитов) – это процесс, включающий 4 стадии созревания. Острый промиелоцитарный лейкоз (ОПЛ) начинается, когда созревающие клетки достигают второго этапа, становятся промиелоцитами и перестают развиваться. Это видно на фото.
Это вызывает сбой, который выражается в следующих процессах:
- Снижение содержания в крови эритроцитов, тромбоцитов, лейкоцитов.
- Замена костного мозга быстро развивающейся опухолью.
Зафиксированы редкие случаи развития ОПЛ при лечении препаратами:
- алкилирующими агентами;
- ингибиторами топоизомеразы II;
- атрациклинами.
Неконтролируемое размножение незрелых клеток вызвано перестройкой 15 и 17-ой хромосом.
Мутация происходит с участием рецептора ретиноевой кислоты и гена, ответственного за деление промиелоцитов. В результате образуется новый ген, который не реагирует на ретиноевую кислоту.
Заболевание характеризуется рядом других генетических изменений, возникающих вследствие описанных нарушений. Эти мутации не влияют на курс оздоровления и смертность.
Распространённость
ОПЛ выявляется у детей и взрослых.
Привязка к возрасту проявляется так:
- 0-17 лет – заболевают 3-4%;
- 10-20 лет – количество постепенно возрастает и останавливается на 5%;
- 20-60 лет – до 10%;
- старше 60 лет – вероятность развития опухоли резко падает.
Ребёнок до 10 лет практически не подвержен развитию опухоли данного вида. Наиболее часто заболевают люди в возрасте 38-40 лет. В Азии распространённость заболевания достигает 69%. Среди жителей Латинской Америки – 73%.
Предполагается, что это обусловлено генетическими особенностями рас.
Риск возникновения промиелоцитарного лейкоза в результате химиотерапии составляет 1,7-5,8%. Чаще избыток промиелоцитов фиксируется после рака молочной железы.
Симптомы
ОПЛ характеризуется скоротечностью. Симптоматика сводится к проявлениям недостатка нормальных кровяных телец и анемии.
- Образуются кровотечения и кровоподтёки из носа, дёсен, в местах незначительного повреждения кожи. Увеличивается менструальное кровотечение у женщин. Образуются синяки.
- Слабость, быстрая утомляемость, одышка, затруднение дыхания.
- Возможно возникновение инфекционных заболеваний из-за сниженного количества лейкоцитов.
ДВС характеризуется внутренними кровоизлияниями из-за разрушения стенок сосудов (геморрагический синдром). Этот процесс происходит при ОПЛ быстрее, чем при ОМЛ. Требуется начать лечение до его появления, чтобы не допустить гибели больного.
Диагностика
Используется комплекс методов для диагностирования ОПЛ.
- Общий клинический анализ крови. Выявляется количество лейкоцитов, тромбоцитов, эритроцитов, гемоглобина. По формуле определяется содержание незрелых клеток.
- Миелограмма – анализ не менее 10 мазков костного мозга.
- Анализ Ликвора – определение цвета костномозговой жидкости. Проводится только при отсутствии кровоизлияний.
- До начала оздоровительных курсов необходимо провести УЗИ брюшной полости, рентгенографию, эхокардиографию и электрокардиограмму.
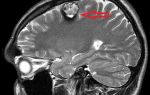
Дополнительно могут быть назначены компьютерная или магнитно-резонансная томография головного мозга (КТ или МРТ). На основании всех анализов устанавливается форма проявления (по величине гранул промиелоцитов) ОПЛ и группа риска (по количеству лейкоцитов до начала терапии).
Для постановки диагноза потребуется молекулярно-генетический анализ. С его помощью выявляется мутирующий транскрипт или транслокация. Без данной процедуры диагноз считается неподтверждённым.
Лечение
Оптимальный курс оздоровления включает транс-ретиноевую кислоту (ATRA). Вещество участвует в сложном процессе взаимодействия хромосом с белками, запускает определённые генные процессы. Результатом становится самоуничтожение лейкозных клеток.
Но продолжительность эффекта при воздействии одной только кислотой крайне мала.
Поэтому её применяют в комплексе по одной из схем:
- ATRA + триоксид мышьяка – считается самой эффективной схемой, и включена в стандарт с 2013 года. При таком курсе, онкологи стараются не применять химиотерапию. Исключением могут стать частные случаи и рецидивы. Триоксид мышьяка менее токсичен, что даёт однозначное преимущество перед химиопрепаратами.
- ATRA + монотерапия атрациклином. Методика считалась стандартной до 2013 г. Сейчас используется в случае невозможности проведения предыдущей.
Воздействие транс-ретиноевой кислоты на организм уникально. Фактически она просто нормализует нарушенные процессы. Но от побочных эффектов это вещество не страхует.
На фоне приёма ATRA развивается ряд несущественных патологий. Все они являются следствием гипервитаминоза витамина А. Также кислота часто вызывает приступы ДВС. Это требует немедленных действий со стороны врача.
Консолидационная программа
Поддержку ремиссии осуществляют в течение 2 лет.
Применяют ATRA в комплексе с цитостатиками:
- меркаптопурин;
- метотрескат.
На базе исследований, проведённых в европейских странах и США, можно сделать вывод, что после установления ремиссии пациенты нуждаются в поддерживающей терапии. По Р.Ф. точной статистики заболеваемости нет. Общая численность лейкозов составляет порядка 5 тысяч случаев в год.
Вероятность рецидива в результате использования курса падает в 2-2,5 раза. В ликвидации повторных очагов развития болезни большая роль отводится триоксиду мышьяка.
Триоксид мышьяка
Исторические сведения указывают на применение этого вещества в качестве яда и лекарственного средства. С начала прошлого века проводились успешные исследования в терапии сифилиса. Десятки лет используется в стоматологии. Первые упоминания о лечении им онкологии относятся к 70 годам ХХ века.
Наиболее значимыми являются американские и китайские исследования в лечении второй линии ОПЛ.
Они показали следующие результаты:
- Китай. 90% случаев вторичных очагов было ликвидировано исключительно мышьяком. У 14 из 15 пациентов наблюдалось полное выздоровление после нескольких рецидивов.
- США. У 6 из 7 больных отмечена полная ремиссия на молекулярном уровне. Однако изначально обследовалось 10 человек, 3 из которых внезапно скончались.
Авторы отмечают, что это могло быть вызвано расовой генетикой или увеличенной дозой препарата.
В целом триоксид оценивается медиками положительно. Выявляются ATRA-подобные побочные эффекты, которые успешно ликвидируются. Отзывы соотечественников о препаратах, используемых в лечении ОПЛ, практически отсутствуют.
Это связано с низкой заболеваемостью в России, а также использованием в стационарах.
Низкая токсичность триоксида мышьяка и естественность процессов при приёме ATRA дают надежду на применение при других формах рака. Проводятся исследования действия этих веществ на различные опухоли.
Промиелоцитарный лейкоз – редкое заболевание, которое поддаётся успешному лечению, хорошо прогнозируется. Однако существуют высокие риски его быстрого развития и внезапного летального исхода. При появлении первых симптомов необходимо обратиться к врачу.
Источник: http://BolezniKrovi.com/rak/krov/ostryy-promielotsitarnyy-leykoz.html
Острый миелоидный лейкоз. Классификация и диагностика

Материал подготовлен специально для сайта НГО: К.А. Матохиной, Е.И. Захарько, Е.Б. Рыбкиной, Д.Г. Дроковой, под руководством Двирнык В.Н. (ЦКДЛ НМИГ гематологии МЗ РФ)
Острый миелоидный лейкоз – это клональное опухолевое заболевание кроветворной ткани, связанное с мутацией в клетке-предшественнице гемопоэза, следствием которой является блок дифференцировки и бесконтрольная пролиферация незрелых миелоидных клеток.
Выделяют 2 основные классификации острых миелоидных лейкозов: FAB-классификация и классификация ВОЗ.
FAB-классификация, предложенная франко-американо-британской (ФАБ) группой экспертов в 1976 г., пересмотренная и дополненная в 1991 г., основывается на морфологической и цитохимической характеристике бластных клеток.
М0 – ОМЛ с минимальной миелоидной дифференцировкой
М1 – ОМЛ без созревания
М2 – ОМЛ с созреванием
М3 – Острый промиелоцитарный лейкоз
М3v – Острый промиелоцитарный лейкоз (гипогранулярный вариант)
М4 – Острый миеломонобластный лейкоз
М4эоз – Острый миеломонобластный лейкоз с эозинофилией
М5а – Острый монобластный лейкоз без созревания
М5в – Острый монобластный лейкоз с созреванием
М6 – Острый эритромиелоз
М7 – Острый мегакариобластный лейкоз
В 2001 году международной группой экспертов была создана новая классификация гемобластозов – классификация ВОЗ, и FAB-классификация стала её составной частью.
Классификация ВОЗ (2016) подразделяет все ОМЛ на клинико-патологические группы, основываясь на их цитогенетических и молекулярно-генетических особенностях.
ОМЛ с повторяющимися генетическими аномалиями:
ОМЛ c t(8;21)(q22;q22);RUNX1-RUNX1T1
Острый промиелоцитарный лейкоз c t(15;17)(q22;q12) и вариантный; PML-RARA
ОМЛ с inv(16)(p13.1q22) или t(16;16)(p13.1;q11);CBFB-MYH11
ОМЛ с t(9;11)(p22;q23);MLLT3-MLL
ОМЛ с t(6;9)(p23;q34);DEK-NUP214
ОМЛ с inv(3) (q21.3.q26.2) или t(3;3) (q21q26.2); GATA2,MECOM
ОМЛ (мегакариобластный) с t(1;22)( p13;q13);RBM15-MKL1
ОМЛ с BCR-ABL1
ОМЛ с мутацией NPM1
ОМЛ с биаллельными мутациями CEBPA
ОМЛ с мутацией RUNX1
ОМЛ, связанный с миелодисплазией
Миелоидные новообразования, ассоциированные с предшествующей терапией
ОМЛ, не охарактеризованные иным образом:
ОМЛ с минимальной дифференцировкой
ОМЛ без созревания
ОМЛ с созреванием
Острый миеломоноцитарный лейкоз
Острый монобластный/моноцитарный лейкоз
Истинный эритролейкоз
Острый мегакариобластныйлейкоз
Острый базофильный лейкоз
Острый панмиелоз с миелофиброзом
Миелоидная саркома
Миелоидные пролиферативные заболевания, ассоциированные с синдромом Дауна:
-транзиторный аномальный миелопоэз (ТАМ)
-ОМЛ, ассоциированный с синдромом Дауна
Диагноз острого миелоидного лейкоза устанавливается при обнаружении в костном мозге ≥20% бластных клеток.
На основании морфологических особенностей бластных клеток различают бласты I типа – арактеризуются высоким ядерно-цитоплазматическим отношением, тонкодисперсным хроматином ядра, наличием одной или нескольких нуклеол и отсутствием азурофильной зернистости в цитоплазме; бласты II типа – отличаются более низким ядерно-цитоплазматическим отношением, наличием азурофильной зернистости и палочек Ауэра (или без них).
Палочки Ауэра – это кристаллические цитоплазматические структуры, образованные из первичных гранул либо сразу после их формирования в цистернах комплекса Гольджи, либо путем объединения гранул внутри аутофагичных вакуолей.
Источник: https://npngo.ru/klassifikatsiya_o
Что такое острый миелобластный лейкоз и какая продолжительность жизни

Острый миелобластный лейкоз (ОМЛ) — термин, который объединяет ряд острых миелолейкозов, характеризующихся развитием сбоев в механизме созревания миелобластов.
На ранних этапах развития болезнь проявляется бессимптомно и диагностируется слишком поздно.
Чтобы выявить лейкоз своевременно, необходимо знать, что это такое, какие симптомы говорят о начале развития заболевания и какие факторы влияют на его возникновение.
Код МКБ-10
Код заболевания — C92.0 (Острый миелобластный лейкоз, относится к группе миелоидных лейкозов)
к содержанию ↑
Что это такое?
ОМЛ — злокачественная трансформация, охватывающая миелоидный росток кровяных клеток.
Пораженные кровяные тельца постепенно заменяют здоровые, и кровь перестает полноценно выполнять свою работу.
Это заболевание, как и другие типы лейкозов, именуют раком крови в повседневном общении.
Слова, из которых состоит это определение, дают возможность понять его лучше.
Лейкоз. При лейкозе измененный костный мозг начинает активно вырабатывать лейкоциты — кровяные элементы, которые ответственны за поддержание иммунной системы — с патологической, злокачественной структурой.
Они замещают собой здоровые лейкоциты, проникают в разные части организма и формируют там очаги поражения, схожие со злокачественными новообразованиями.
Отличия здоровой крови от больной лейкозом
Миелобластный. При ОМЛ начинается избыточное продуцирование пораженных миелобластов — элементов, которые должны превратиться в одну из разновидностей лейкоцитов.
Они вытесняют здоровые элементы-предшественники, что приводит к дефициту других кровяных клеток: тромбоцитов, эритроцитов и нормальных лейкоцитов.
Острый. Это определение говорит о том, что продуцируются именно незрелые элементы. Если пораженные клетки находятся в зрелом состоянии, лейкоз называется хроническим.
Лейкозк содержанию ↑
Симптомы
Обычно ОМЛ развивается у взрослых и пожилых людей. Ранние стадии миелобластной лейкемии характеризуются отсутствием ярко выраженной симптоматики, но, когда заболевание охватило организм, возникают серьезные нарушения многих функций.
к содержанию ↑
Гиперпластический синдром
Развивается из-за тканевой инфильтрации под действием лейкоза. Разрастаются периферические лимфоузлы, увеличивается селезенка, нёбные миндалины, печень.
Кровоток в ней нарушается, что сопровождается возникновением отечности в зоне шеи, учащенного дыхания, синюшности кожных покровов, набуханием сосудов на шее.
Также поражаются десна: появляется стоматит Венсана, который характеризуется развитием тяжелых симптомов: десны отекают, кровоточат и сильно болят, есть и ухаживать за полостью рта затруднительно.
к содержанию ↑
Геморрагический синдром
Более половины больных имеют те или иные его проявления, развивается из-за острой нехватки тромбоцитов, при которой стенки сосудов истончаются, нарушается свертывание крови: наблюдаются множественные кровотечения — носовые, внутренние, подкожные, которые долгое время не удается остановить.
Увеличивается риск геморрагического инсульта — кровоизлияния в мозг, при котором летальность составляет 70-80%.
к содержанию ↑
Анемия
Характеризуется появлением:
- Выраженной слабости;
- Быстрой утомляемости;
- Ухудшением трудоспособности;
- Раздражительности;
- Апатии;
- Частых болей в голове;
- Головокружений;
- Обмороков;
- Стремления есть мел;
- Сонливости;
- Болей в области сердца;
- Бледности кожи.
к содержанию ↑
Интоксикация
Температура тела повышена, вес падает, аппетит пропадает, наблюдается слабость и избыточное потоотделение.
Начальные проявления интоксикации наблюдаются на начальных этапах развития заболевания.
к содержанию ↑
Нейролейкоз
Если инфильтрация затронула мозговые ткани, это ухудшает прогноз.
Наблюдается следующая симптоматика:
- Многократная рвота;
- Острая боль в голове;
- Эпиприступы;
- Обмороки;
- Внутричерепная гипертензия;
- Сбои в восприятии реальности;
- Нарушения слуха, речи и зрения.
к содержанию ↑
Лейкостазы
Развиваются на поздних стадиях заболевания, когда количество пораженных миелобластов в крови становится выше 100000 1/мкл.
Кровь густеет, ток крови становится медленным, нарушается кровообращение во многих органах.
Мозговой лейкостаз характеризуется возникновением внутримозговых кровотечений. Нарушается зрение, возникает сопорозное состояние, кома, возможен летальный исход.
При легочном лейкостазе наблюдается учащенное дыхание (возможно возникновение тахипноэ), озноб, повышение температуры. Количество кислорода в крови сокращается.
к содержанию ↑
Причины
Точные причины развития ОМЛ неизвестны, но существует ряд факторов, которые увеличивают вероятность развития заболевания:
- Радиационное облучение. В группе риска люди, которые взаимодействуют с радиоактивными материалами и приборами, ликвидаторы последствий ЧАЭС, пациенты, проходящие лучевую терапию при другом онкологическом заболевании.
- Генетические заболевания. При анемии Факони, синдромах Блума и Дауна риск развития лейкоза увеличивается.
- Воздействие химических веществ. Химиотерапия при лечении злокачественных заболеваний негативно воздействует на костный мозг. Также вероятность повышается при хроническом отравлении ядовитыми веществами (ртуть, свинец, бензол и прочие).
- Наследственность. Люди, близкие родственники которых страдали лейкозами, также могут заболеть.
- Миелодиспластический и миелопролиферативный синдромы. Если лечение одного из этих синдромов будет отсутствовать, заболевание может трансформироваться в лейкоз.
к содержанию ↑
Формы ОМЛ
Миелобластная лейкемия имеет ряд разновидностей, от которых зависит прогноз и тактика лечения.
| ОМЛ с незначительной дифференциацией (М0). | Низкая восприимчивость к химиотерапевтическому лечению, легко приобретает резистентность к ней. Прогноз неблагоприятный. |
| ОМЛ без созревания (М1). | Отличается стремительным прогрессированием, бластные клетки содержатся в большом количестве и составляют порядка 90%. |
| ОМЛ с созреванием (М2). | Уровень моноцитов при этой разновидности — менее 20%. Не меньше 10% миелобластных элементов развиваются до стадии промиелоцитов. |
| Промиелоцитарный лейкоз (М3). | В костном мозгу интенсивно накапливаются промиелоциты. Относится к наиболее благоприятным по течению и прогнозу лейкозам — в течение 10-12 лет живут не менее 70%. Симптоматика схожа с остальными разновидностями ОМЛ. Лечится с применением оксида мышьяка и третиноина. Средний возраст заболевших — 30-45 лет. |
| Миеломоноцитарный лейкоз (М4). | Диагностируется у детей чаще, чем другие разновидности заболевания (но в целом ОМЛ в процентном соотношении, по сравнению с другими типами лейкозов, выявляется у детей редко). Лечится с применением интенсивной химиотерапии и пересадки стволовых клеток (ТГК). Прогноз неблагоприятный — показатели выживаемости в течение пяти лет — 30-50%. |
| Монобластный лейкоз (М5). | При этой разновидности в костном мозгу содержится не менее 20-25% бластных элементов. Лечится химиотерапией и ТГК. |
| Эритроидный лейкоз (М6). | Редко встречающаяся разновидность. Лечится с применением химиотерапии и пересадки стволовых клеток. Прогноз неблагоприятный. |
| Мегакариобластный лейкоз (М7). | Этой разновидности ОМЛ подвержены люди с синдромом Дауна. Характеризуется быстрым течением и низкой восприимчивостью к химиотерапии. Детские формы болезни чаще текут благоприятно. |
| Базофильный лейкоз (М8). | Чаще встречается в детском и юношеском возрасте, прогноз жизни М8 неблагоприятный. Помимо злокачественных элементов, в крови выявляются аномальные элементы, которые затруднительно выявить без специального оборудования. |
к содержанию ↑
Диагностика
Острая лейкемия выявляется с применением ряда диагностических мероприятий.
Диагностика включает в себя:
- Развернутый анализ крови. С его помощью выявляется содержание в крови бластных элементов и уровень остальных кровяных телец. При лейкемии обнаруживается избыточное количество бластов и сниженное содержание тромбоцитов, зрелых лейкоцитов, эритроцитов.
- Взятие биоматериала из костного мозга. Применяется для подтверждения диагноза и проводится после проведенных обследований крови. Этот метод применяется не только в процессе диагностики, но и на протяжении лечения.
- Биохимический анализ. Дает информацию о состоянии органов и тканей, содержании различных ферментов. Этот анализ назначается для получения развернутой картины поражения.
- Другие виды диагностики: цитохимическое исследование, генетическое, УЗИ селезенки, брюшной полости и печени, рентген зоны груди, диагностические мероприятия для выявления степени поражения головного мозга.
к содержанию ↑
Лечение
Лечение ОМЛ включает применение следующих методов:
- Химиотерапия. Медикаменты воздействуют на клетки, подавляя их активность и размножение. Ключевой метод лечения лейкемии.
- Консолидация. Лечение, назначаемое во время ремиссии, нацелено на снижение вероятности рецидива.
- Трансплантация стволовых клеток. Пересадка костного мозга при лейкозе применяется при лечении больных младше 25-30-летнего возраста и необходима в тех ситуациях, когда заболевание течет неблагоприятно (наблюдается нейролейкоз, концентрация лейкоцитов крайне высока). Пересаживаются либо собственные клетки, либо донора. Обычно донорами становятся близкие родственники.
- Дополнительная терапия. Восстанавливает состояние крови, включает введение кровяных элементов.
Применяются:
- Медикаменты на основе моноклональных антител;
- Адаптивная клеточная терапия;
- Ингибиторы контрольных точек.
к содержанию ↑
Прогноз жизни
Прогноз зависит от следующих факторов:
- Типа ОМЛ;
- Чувствительности к химиотерапии;
- Возраста, пола и состояния здоровья пациента;
- Уровня лейкоцитов;
- Степени вовлеченности головного мозга в патологический процесс;
- Продолжительности ремиссии;
- Показателей генетического анализа.
Если заболевание чувствительно к химиотерапии, концентрация лейкоцитов умеренная, а нейролейкоз не развился, прогноз положительный.
При благоприятном прогнозе и отсутствии осложнений выживание в течение 5 лет составляет более 70%, частота рецидивов менее 35%. Если состояние пациента осложнено, то выживаемость равняется 15%, при этом рецидивировать состояние может в 78% случаев.
к содержанию ↑
Видео: Острый миелолейкоз
Источник: https://moyakrov.info/blood/lejkoz/ostryj-mieloblastnyj
Острый лейкоз крови

Острый лейкоз не без оснований приравнивают к злокачественным образованиям. Действительно, болезнь вызвана чрезвычайным разрастанием молодых (бластных) клеток крови. Но, в отличии от опухолевых, они не являются атипичными по строению или чужеродными для организма. Это «родные» клетки, которые вдруг начинают синтезироваться в большом количестве.
В зависимости от морфологии начальных элементов различают 2 вида острых лейкозов: лимфобластный и миелобластный.
Медицинская статистика установила:
- общая заболеваемость составляет 35 случаев на млн. населения;
- наиболее ранимые возрастные периоды заболевания: детство и у взрослых старше 40 лет;
- женщины и мужчины заболевают с разной частотой, распространенность среди женщин – 7,7 на 100 тыс., среди мужчин — 13,2;
- соотношение между лимфобластными и нелимфобластными формами меняется в зависимости от возрастных групп, среди детей до 15 лет лимфобластных больше в 4 раза (4:1), а среди людей старше 35 лет миелобластные выявляются в 8 раз чаще (1:8).
Причины лейкоза крови до настоящего времени не выявлены. Большинство ученых считают главной — изменения на уровне строения и состава хромосом.
Как развивается болезнь
Лишние бластные клетки разрастаются и занимают место нормальных элементов крови, вытесняя их и подавляя правильное кроветворение.
Под влиянием лечения цитостатиками происходит приспособление и прогрессирующий рост новых клональных клеток, из которых далее развиваются бласты.
Метастазы бластов возникают в селезенке, лимфоузлах, печени (органах, принимающих участие в кроветворении,) и других частях тела (головной мозг, кожа, легкие, яички).
Классификации
Длительное время ученые разных стран и клиницисты пользовались классификацией (FAB), основанной на «внешнем виде» основных клеток: размеры, соотношение между массой ядра и цитоплазмы, строение и форма ядра. Доводы против этого вида учета предъявляли практические гематологи, занимавшиеся диагностикой и лечением.
Классификация не позволяла ориентироваться в разновидностях патологии при выборе средств терапии, определении стандартов тактики в лечении, не учитывала прогноз заболевания
Поэтому Всемирная организация здравоохранения в 2001 году приняла новую более удобную классификацию. К названиям формы заболевания добавляется код поврежденного хромосомного набора, понятный для ученых-генетиков.
Виды острых лимфобластных лейкозов
Современная классификация острых лейкозов лимфобластного типа строится на определении маркеров иммунных клеток (специфических антигенов к определенным клонам):
- если на поверхности клеток обнаруживаются маркеры к Т-лимфоцитам, Т- острый лимфобластный лейкоз;
- при родстве к В-ряду лимфопоэза — В-лимфобластный;
- общая или смешанная форма заболевания.
В группах выделены подвиды, зависящие от стадий развития (дифференциации) клеток:
- пре -Т, про-Т, Т зрелый;
- пре -В, пре-пре-В, про-В.
Отдельно выделены виды, сопровождающиеся известными хромосомными аномалиями:
- с Филадельфийской хромосомой;
- с транслокациями (заменой) одной хромосомы на другую и образованием нового гена.
Виды острых нелимфобластных лейкозов
Формы нелимфобластных лейкозов с генетическими аномалиями тоже учитывают известную замену местами пронумерованных хромосом. Подобных классов выделено 4. Кроме того, имеются подвиды:
- с изменениями всех рядов клеток (мультилинейной дисплазией);
- лейкоз на фоне другого предшествующего заболевания (прелейкемического), сопровождающегося разрастанием или изменением структуры клеток нелимфатического ряда;
- заболевание без доказанного прелейкемического периода, но при обнаруженном нарушении строения более половины клеточного состава по разным формам дифференцировки;
- вторичный острый лейкоз, появившийся на фоне лечения онкологического заболевания методом химиотерапии.
К очень редким подвидам относятся выделяемые в детском возрасте:
- базофильная форма лейкоза,
- острый панмиелоз и миелофиброз,
- саркома миелоидного характера.
Старая (морфологическая) классификация
Если по морфологическим характеристикам форма лейкоза не входит в классификацию ВОЗ, то в диагностике пользуются старыми названиями и видами.
Франко-американо-британская разновидность (FAB) предлагает учет острых нелимфобластных лейкозов по анализам крови и клеткам костного мозга. Диагностика формы основывается на количестве клеток, процентном составе. Для краткости каждому виду присвоено буквенное изображение с порядковым номером (М1, М2).
М1 — острый миелоидный лейкоз (миелобластный):
- в костномозговом веществе бластных клеток не меньше 90% состава;
- остальные 10% приходятся на созревающие гранулоциты (от промиелоцитов до сегментоядерных лейкоцитов).
М2 — разновидность М1, но клетки характеризуются как «частично созревшие»:
- бластных клеток от 30 до 90%;
- менее 20% клеток моноцитарного ростка;
- гранулоцитов — 10% и более.
М3 — острый промиелоцитарный лейкоз диагностируется при обнаружении в содержимом костного мозга промиелоцитарных клеток без дополнительных тестов.
М4 — острый миеломонобластный лейкоз:
- в костном мозге содержится от 30 до 80% клеток миелоидного ростка;
- в периферической крови моноцитов и их предшественников 5х109 и более в литре.
Кроме структуры клеток, используют реакцию окраски бластных клеток с α-нафтилэстеразой (положительный и отрицательный варианты).
При одновременном содержании в крови эозинофилов не менее 5% форма заболевания называется М4э.
М5 — острый монобластный лейкоз: монобласты в пунктате составляют не меньше 80%.
М7 — острый мегакариобластный лейкоз: в диагностике необходимо иммунофенотипирование или электронная микроскопия.
М0 — острая ранняя миелобластная форма: все окраски дают отрицательный результат, для диагностики необходимо изучение иммунного типа клеток.
Клинические проявления
Острый лейкоз проявляется клиническими симптомами, независимо от вида заболевания. Начало болезни может быть внезапным или развиваться постепенно.
Нарушенная работа костного мозга вызывает клинические синдромы:
- анемический — у пациента наблюдается бледность кожи, жалобы на общую слабость, сонливость, сердцебиение;
- геморрагический — на коже самопроизвольно появляются кровоподтеки в местах сдавления, трения, инъекций, мелкие точечные кровоизлияния (петехии), пациент страдает от носовых кровотечений, у девушек и женщин возможны обильные менструации, в тяжелых случаях возникают проявления желудочного и кишечного кровотечения, кровоизлияния в мозг;
- развитие иммунодефицита способствует быстрому заражению любыми инфекциями (вирусными, бактериальными), особенно тяжело они протекают, если присоединяется грибковая флора, у пациентов обнаруживают стоматиты, герпетические высыпания, ангины, в тяжелых случаях воспаление легких, сепсис;
- синдром диссеминированного внутрисосудистого свёртывания (ДВС), обычно вызывает острый промиелоцитарный лейкоз, свертываемость крови проходит стадии повышения и резкого снижения, усиливаются кровотечения.
При носовых кровотечениях анемия еще более усиливается
Специфические признаки
Острый лейкоз имеет ряд специфических признаков:
- Проявления общей интоксикации — повышение температуры, усиление потливости, снижение аппетита, похудение, слабость.
- Боли в костях позвоночника, больших трубчатых.
- Увеличение лимфоузлов в любой зоне, узлы при пальпации плотные, круглые, эластичные, могут быть групповыми, спаянными между собой, болезненные.
- Локализация увеличенных узлов в области брыжейки вызывает боли в животе. Если увеличиваются загрудинные лимфоузлы, то возможно сдавление бронха, пищевода. Соответственно, появляется нарушение дыхания, глотания.
- Поражение головного мозга (нейролейкемия) — чаще наблюдается при лимфобластной форме, симптомы связаны с метастазами клеточных разрастаний в оболочки и вещество головного и спинного мозга, наблюдаются головные боли, головокружения, ухудшение зрения, изменение речи, координации движений.
- Лейкемиды (узелки) на коже чаще дает острый миелоидный лейкоз, миеломонобластная форма.
- Значительное увеличение вилочковой железы (тимуса) способствует сдавлению органов средостения.
- Поражение яичек проявляется в ассиметричном увеличении, отсутствии болей.
Диагностика
Первые изменения после клинических симптомов отмечают в анализе крови. Любые резкие отклонения в формуле при нормальном, пониженном или повышенном общем количестве лейкоцитов вызывают подозрения на лейкоз.
Дифференциация острой или хронической форм зависит от наличия молодых клеток на фоне отсутствия промежуточных (лейкемический провал). Почти всегда имеется лимфоцитоз, тромбоцитопения, анемия.
Основа диагностики — цитологическая характеристика пунктата костного мозга, определяются характерные описанные признаки в миелограмме.
Цитохимический метод основан на выявлении типичных для разных бластных форм ферментов.
Методика иммунофенотипирования позволяет типировать бластные клетки по характерным маркерам. Она необходима в случаях затруднения отличия лимфобластного и миелобластного лейкоза. Это важно, поскольку требуется разное лечение.
Также почитать:Причины острого лимфобластного лейкоза
Генетические исследования клеток крови позволяют установить патологию хромосом, конкретную генетическую аномалию.
Анализ спинномозговой жидкости после пункции имеет значение в диагностике степени поражения мозга.
На рентгеновском снимке грудной клетки можно увидеть увеличенные группы лимфоузлов в легких, средостении.
Перед применением химиотерапии врачу необходимы исходные показатели работы печени и почек (биохимия крови), электрокардиограмма, УЗИ печени, селезенки. По ним будет оцениваться токсический эффект лечения.
Лечение
Лечение острого лейкоза проводится в специализированных отделениях. Начинать его следует незамедлительно. Главная цель терапии — достижение длительной ремиссии болезни, когда пациент не чувствует симптомов, стабилизируется картина крови.
Современные методы терапии включают:
- химиотерапию,
- биологический способ,
- лучевой метод,
- пересадку стволовых клеток костного мозга донора,
- удаление селезенки по показаниям (в поздних стадиях).
Химиотерапия
Метод химиотерапии предполагает использование сильных лекарственных средств, способных разрушить измененные клетки. Препараты вводятся внутривенно, в спинномозговой канал.
В связи с необходимостью частых внутривенных вливаний используются приспособления для многократных инфузий
Применяется курсовая терапия с расчетом общей дозы препаратов на вес больного. Подбор препаратов и стандартного курса определяется по степени нарушения кроветворения.
После стационарного лечения больному назначают поддерживающую дозировку для приема на дому.
Биометод
К биологическому методу относят стимуляцию собственного иммунитета организма для торможения роста раковых клеток. С этой целью используют моноклональные антитела, Интерферон.
Лучевое воздействие
Лучевая терапия — воздействие радиацией на костный мозг больного. Врачи устанавливают место для облучения, разовую и курсовую дозу. Часто используется перед пересадкой костного мозга.
Новейшее оборудование для лучевой терапии позволяет минимизировать воздействие на здоровые ткани
Трансплантация костного мозга
Пересадка здоровых стволовых клеток костного мозга позволяет повысить дозу химиопрепаратов и лучевой терапии. Из пересаженных клеток развиваются нормальные лейкоциты.
Нежелательные эффекты лечения
Перечисленные методики не обходятся без осложнений. Важно, что они ожидаемы и знакомы докторам. Пациентам тоже рассказывают об отрицательных последствиях терапии.
При химиотерапии наступает: кровоточивость, усиливается слабость из-за малокровия, облысение. На губах появляются язвочки, поражается слизистая кишечника (тошнота, потеря аппетита, рвота).
После биологических методик возможно появление сыпи на коже, зуда, гриппоподобного состояния с высокой температурой.
При курсе радиационной терапии пациент чувствует повышение слабости, кожа сохнет, стареет, краснеет.
Трансплантация не всегда бывает успешной: если развивается реакция отторжения, возникает непосредственная угроза для всех внутренних органов, особенно для печени.
Поддерживающая терапия
В состоянии ремиссии лечение пациента не прекращается, ему показана поддерживающая терапия и симптоматические средства для профилактики рецидивов болезни. Используются антибактериальные препараты, переливание крови, витамины. Все средства назначаются только врачом.
Прогноз
Прогноз определяется по показателю пятилетней выживаемости пациентов. При остром лимфобластном лейкозе он в настоящее время достиг 68,8% у взрослых, при миелобластном — 24,9%.
При лечении острого лейкоза показатели у детей значительно лучше: 85% в случаях острого лимфобластного заболевания, от 60 да 70% – при миелобластной форме.
Считается, что можно говорить о клиническом выздоровлении детей, перенесших острую форму лейкоза.
Самыми неблагоприятными формами пока являются: острый лимфолейкоз с Филадельфийской хромосомой, нелимфобластные формы с множественной дисплазией клеток. Описанные варианты – М2, М3 и М4 – отличаются хорошей реакцией на химиотерапию.
Успехи в лечении детского лейкоза позволяют надеяться на получение новых средств и достижение длительной ремиссии при заболеваниях взрослых.
Источник: http://serdec.ru/bolezni/ostryy-leykoz-krovi